 |
Constraints & Restraints
III. Thermal Motion |
|
|
Constraints & Restraints
III. Thermal Motion |
Thermal Motion Constraints
Anisotropic Temperature Factors
At very low temperatures, atoms usually vibrate equally in all directions, so isotropic temperature factors are often adequate to describe the smearing out of the scattering density of the atoms. At higher temperatures, anisotropic displacement parameters (ADPs) are normally required in the refinement of a crystal structure. In contrast to the isotropic case, the point group symmetry of the atom determines any symmetry constraints that have to be applied to the 6 anisotropic temperature factors, βij.
The effect of symmetry on anisotropic temperature factors can be illustrated using the two diagrams below:

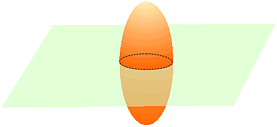
In the left-hand figure, the thermal ellipsoid is tilted at an angle to the plane shown in light green. If this plane were to have mirror symmetry, then application of symmetry to the solid ellisoid would give the ellipsoid outlined with the dashed black lines. The latter does not superimpose onto the original and will break the symmetry of the mirror plane if left as is. The only way in which superposition can take place is when one of the axes of the ellisoid is perpendicular to that of the plane. This is shown in the right-hand figure. Assuming that the mirror plane is perpendicular to the c axis of the unit cell, then the symmetry constraints applied to the temperature factors for this atom are:
This particular symmetry constraint was used in the Rietveld case study described previously for those atoms in the structure of lead sulphate that lay on the mirror planes in space group Pbnm. Note that similar symmetry constraints are also applied to B or U values if the latter are used instead of β. Modern Rietveld programs should have these constraints built into the refinement procedure, but alas many do not!
Mathematically, temperature factor constraints can be determined by testing the invariance of the temperature factor tensor matrix, U, with respect to the point-group symmetry operations of the site, i.e.
| Site Symmetry | Constraint | |
|---|---|---|
| -1 | (none) | |
| m | β12 = β13 = 0 | |
| m | β12 = β23 = 0 | |
| m | β13 = β23 = 0 | |
| 222, mm2, or mmm | β12 = β13 = β23 = 0 |
With atoms positioned on sites of very high symmetry, more extreme constraints
may be required. The ultimate temperature factor constraint is for atoms
with point-group symmetry m-3m, e.g. the Na+ and Cl- ions in
the rock salt structure, where the symmetry constraint is
Whole Body Molecular Motion
The use of anisotropic temperature factors for every atom in a Rietveld refinement dramatically increases the number of refined parameters compared to isotropic case. In many cases, the quality of the data may not support the number of parameters being refined. There are several solutions to this. The first involves the use of rigid bond restraints in which the thermal displacement of two atoms along the line joining them is restrained to be equal. This feature is rarely available in Rietveld programs.
The second solution, which is used more commonly in protein crystallography is to treat the whole molecule (or suitable parts of it) as a rigid body. The thermal motion of a rigid body can be described in terms of a translational tensor, T, which is analogous to the U tensor for the anistropic displacement of individual atoms, a librational tensor, L, to describe whole body vibration about a point, and a screw tensor, S, which corresponds to a combination of translation and rotation, i.e. screw motion. The use of these three tensors is referred to as a TLS refinement. When the site symmetry of the molecule (or group) has a centre of symmetry, the matrix S is zero reducing the number of parameters even further. Again, this option is rarely coded into many Rietveld programs.
|
© Copyright 1997-2006.
Birkbeck College, University of London.
|
Author(s):
Jeremy Karl Cockcroft
Simon Jacques |