 |
Principles |
|
|
Principles |
Principles
What is Indexing?
Indexing is the process by which reflection indices, hkl, are assigned to the peaks in the diffraction data. As soon as a sufficient number of reflection indices have been assigned to particular peaks, then the unit cell can be calculated (or refined) as discussed in the previous section on unit-cell refinement. Compared to indexing the peaks from single-crystal data (which have the 3 angle parameters ω,χ,φ) the process of indexing a powder diffraction pattern is much harder since the 3-dimensional information is reduced to a 1-dimensional set of d spacings.
In order to calculate the position of all of the peaks in a powder pattern, 6 unit-cell parameters (a, b, c, α, β, γ) and a wavelength are the minimum requirements: so how many peaks are required for the reverse procedure? The answer is not so obvious!
Suppose we have a cubic powder diffraction pattern and we know that the first peak is a 111 reflection (say from a face-centred cubic structure), then the answer might be 1 since from this information we can obtain the unit cell. However, although unit cells with cubic symmetry have only a single parameter, we still need to have more than one peak in order to know with some certainty that the pattern really does correspond to that of a cubic material. Assuming that the symmetry of the unit cell is unknown, then obviously 6 unit-cell parameters are to be determined: in practice, the number of peaks required is approximately 3 times this number and most automatic indexing programs for powders require a minimum of 20 reflections as input data.
Data Collection Strategy
Data acquisition for indexing purposes is quite different to that collected for, say, structure refinement where good statistics for peaks with small d-spacing values are important. Firstly, given that a limited number of reflections are required for indexing, then there is no need to collect the whole powder diffraction pattern. Secondly, given the importance of large d-spacing values, then these should be measured to the best possible accuracy as regards peak position.
By contrast, accurate peak intensities are often irrelevant (at least to a first approximation): this permits silicon wafer sample holders to be used for organic materials, thus obtaining sharp and precise peaks for materials with high transparency to X-rays. However, it is important that peaks are measured with sufficient intensity so that their position can be obtained reliably. In some cases, it may be worthwhile to measure weak peaks more than once so as to obtain good intensity statistics.
One useful trick is to measure the powder pattern in both reflection and transmission geometries (if this option is available). Sometimes very weak peaks may show up when measured with one geometry, but not the other. In extreme cases, the characteristic spacing of layered systems may be evident from preferred orientation, and this may be an aid to indexing.
Q Space
In the previous section on unit-cell refinement, it was shown that the d spacing of a particular reflection with indices hkl is given by:
where A to F are 6 parameters which are directly related to the reciprocal unit cell. For indexing purposes, it is therefore useful to treat the data as a set of 1/d2 values. Working in Ångstrom units, d is usually much bigger than 1; hence it is common to multiply the 1/d2 values by 104 so as to avoid small numbers. These values are sometimes referred to as "Q" values:
They should not to be confused with
the Q values conventionally used in
scattering theory, especially neutron scattering theory, which are
defined differently, and which are equal to
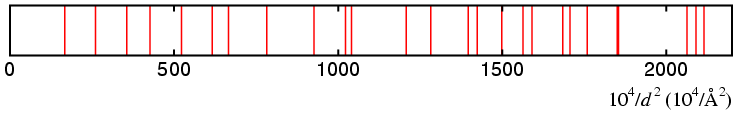
All indexing programs have to decide whether a line is indexed or not since all measured observations intrinsically possess some kind of error. Usually some tolerance is set. For instance, in the case of angle-dispersive data a line may be defined as being indexed if the calculated and observed 2θ values differ by less than 0.05°. Clearly the acceptance value depends on the resolution of the powder diffractometer on which the data were collected.
Now a solution is more likely to "index" an observed line if the solution generates lots of hkl triplets. The number of reflections from a single crystal, N(hkl), is given approximately by the formula:
where V is the real-space volume of the unit cell. In a powder pattern there will be many coincidences or overlaps between reflections with distinct hkl's but equal or nearly equal d spacings, which will reduce the number of peaks apparent in the diffraction pattern. Despite this, the number of peaks is still proportional to V. Thus a solution with double the unit-cell volume will have twice as many hkl values and double the probability of indexing a line by chance. It is therefore not only desirable, but essential, to have some means of distinguishing different solutions automatically.
Figure of Merit, M20
The most widely used figure of merit to assess the quality of the solutions provided by indexing programs is the value M20 devised by de Wolff, which is defined as follows:
where N(calc) is the number of hkl generated with Q values less than or equal to the Q value of the 20th observed and indexed line in the powder pattern, Q20(obs). The quantity <δ> is the mean discrepancy between observed and calculated 2θ values for the lines. This definition has resulted in the use of a minimum of 20 observed powder lines as main data input to most of the powder indexing programs in use today.
How are the values of M20 to be interpreted? One suggestion quoted from the ITO program by J.W. Visser is:
In the above formula that related V to the number of reflections it was assumed that the unit cell is primitive. If a crystallographer chooses a centred unit cell by reason of symmetry, then an additional dividing factor, n, must be included to reduce the number of calculated reflections: for A, B, C, F, I, and R centred lattices, the value of n is 2, 2, 2, 4, 2, and 3, respectively.
![]() You can test the validity of the above statement using the
You can test the validity of the above statement using the
![]() generating program.
generating program.
| © Copyright 1997-2006. Birkbeck College, University of London. | Author(s): Jeremy Karl Cockcroft |