 |
Using Rietica: III. Getting Started |
|
|
Using Rietica: III. Getting Started |
Getting Started
Let us assume that you are starting the refinement of a crystal structure from scratch. Once you have checked that the data appears sensible and have set up the instrumental preferences, you are now in a position to start the analysis. The first step with Rietica is to create a new crystallographic information file, which will have the extension ".inp". On the main menu bar, pull-down the File menu and click on New Input, as shown next:
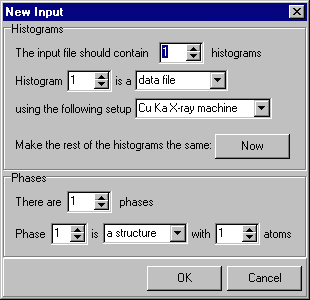
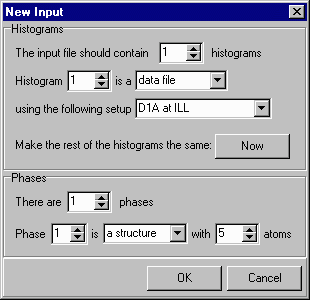
Since this demonstration involves the refinement of lead sulphate from powder neutron diffraction data, you need to change the instrument to, say, 2 and, in addition, set the number of atoms to 5 (as there are five atoms in the asymmetric unit: Pb, S, O(1), O(2), & O(3)), as shown here:
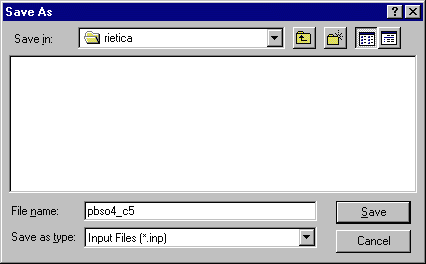
When you click on OK, you will then be prompted for a filename for the new refinement crystallographic input file. It is a good idea to make this filename the same as the data filename (but without any extension) so that Rietica will automatically find the data associated with it:
Click on
You are now in a position to enter information about the crystal structure
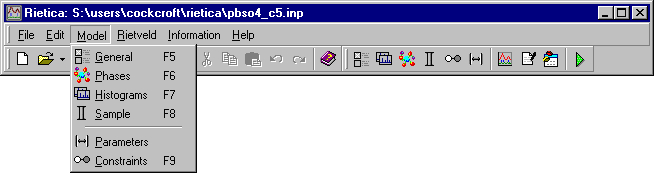
and its refinement by going to the Model pull-down menu,
which has several options as shown below:
At this point you could begin entering values for the
initia coordinates, etc.,
of your crystallographic model. However, it is more logical to start with
the background of the diffraction pattern, which will be described next.
![]() Course Material Index
Course Material Index
![]() Section Index
Section Index
![]() Previous Page
Previous Page
![]() Next Page
Next Page
© Copyright 2001-2006.
Birkbeck College, University of London.
Author(s):
Jeremy Karl Cockcroft
Lachlan M.D. Cranswick